Because they carry both electricity and ions, hybrid organic-inorganic perovskites (HOIPs) are an important type of material in the field of photovoltaics. They use both electrons and ions to move charges around. Ions move by hopping between thermodynamically favourable ion gaps, while electrons move using the band transport process. When electrons and ions move back and forth, it can cause problems like hysteresis in current-voltage curves, thermoelectricity, switchable solar effects, sensitivity to heat and light, and better power conversion rates. To make new HOIPs with less hysteresis, better stability, and better device performance for energy gathering and other electrical uses, it is important to understand the link between electronic and ionic conductivity. The point of this study is to figure out how ions move through perovskites when they are affected by outside forces and how that affects the device’s overall conductivity and performance. It is talked about how device performance changes when there are outside problems, and stable issues with HOIPs devices are brought up.
Vacancy-Mediated Ion Transport (Microscopic
Transport Mechanism of Ion Migration)
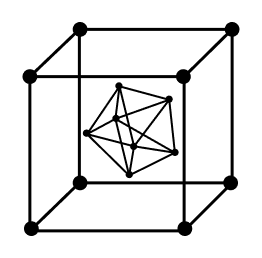
Ionic conductivity can be broken down into two types: the movement of flaw sites and the movement of different types of ions. The movement of ionic species is also caused by defect sites. This has big effects on the performance and long-term security of perovskite-based products. The movement of natural ionic vacancies and interstitial flaws in organolead halide perovskites is very important for these devices. The vacancy-mediated ion transport process that was seen under a microscope shows mixed ionic and electronic conduction in organic–inorganic hybrid perovskites.
Most of the defects that can happen in organolead halide perovskites are point defects, such as ionic vacancies (VMA, VPb, and VI) and interstitials (MAi, Pbi, and Ii). There are also some cation substitutions (MAPb and PbMA) and antisite substitutions (MAI, PbI, IMA, and IPb). Most likely, these flaws are caused by their low formation energies. They let ions move around in the material by making small donor and acceptor levels close to the band ends.
The vacancy-mediated ion transport works better in CH3NH3PbI3 because it’s easy for these Schottky disorders to form, and there are more than 0.4% natural vacancies at room temperature. The following reaction in Kröger–Vink notation shows how these Schottky- and Frenkel-type ionic disorders with vacancies and interstitials are stronger than the electronic disorder in CH3NH3PbI3. Differences in the crystal structure caused by two other ions are not shown here because they are too big to fit in the spaces between the atoms of CH3NH3PbI3.
Vacancy-Mediated Ion Transport Mechanism
Four basic defects have been used to describe how flaws move across the perovskite crystal. The movement path involves normal hopping from one good place to another nearby. For VI, I-ions move along an octahedron edge from the axial site to the equatorial position. This causes the void VI to move from the equatorial position to the axial site. The activation energy of VI is the lowest compared to VMA and VPb, which means that VI-dominated defect migration is most likely to happen in CH3NH3PbI3 perovskites.
For VMA, the MA+ ion moves from one cavity to the next, taking up a spot in the next empty A-site. The activation energy of MA+ is higher than that of I-ion. This means that MA+ ions move slowly and need to be disturbed from the outside for them to move significantly in the perovskite material.
For VPb migration, an in-plane migration is looked into. VPb moves along the side of the face square of cubic unit cells made up of four Pb2+ ions at the corners and I− ions at the edge centres that are tilted a little. The high energy barrier for VPb migration (0.80–2.50 eV) points to a Pb sublattice that is not moving.
Ii moves along the c-axis between interstitial sites in the crystal lattice that are next to each other. The active energy of Ii migration is the same as that of VI migration. This means that Ii migration and VI migration are stronger than VMA and VPb migrations in perovskite crystals that are high in I.
The diffusion value of I− ions is about four orders of magnitude higher than that of MA+ ions. This suggests that in CH3NH3PbI3 perovskite crystals, iodine ions move more easily than methylammonium ions. These findings show that hybrid organic–inorganic perovskite materials have mixed ionic and electronic conductance.
Paths of Vacancy Migration
Depending on where the ions start and end their journey, perovskite crystals have more than one way for them to move. These routes are the same for iodine and methylammonium ions in a cubic perovskite crystal structure and have the same amount of energy. But in tetragonal and trigonal crystal structures, all movement routes are not the same because of the way the structures are set up. Iodine and methylammonium ions can move through the CH3NH3PbI3 crystal’s tetragonal lattice structure in a number of different ways.
(I−) ion migration pathways
Iodine ions are placed at the six corners of the PbI6 octahedron in the perovskite crystal structure. There are two types of iodine ions: those that are equatorial and those that are axial. P1: from axial site to axial site, P2: from axial site to equatorial site, and P3ʈ: from equatorial site to equatorial site. These are the three ways that iodine could move across the octahedron. It would take a lot of energy for the third path to be able to cross the twice positively charged Pb ion, so it is not possible.
For a tetragonal perovskite crystal, the only lines that can work are P1 and P2. Path P1 migration goes in the direction of 111ˀ and path P2 migration goes in the direction of 100ˀ. More activation energy is needed along path P2 than along path P1, because movement along path P1 is between similar points in the same plane and not as far. However, movement along path P2 changes the position of the iodine in the octahedron and covers a longer distance, which makes path P1 better.
In the cubic crystal structure of CH3NH3PbI3, the energy levels of the axial and equatorial places in the octahedron are almost the same. This means that both paths are isoenergetic and equal.
(MA+) ion migration pathways
The process of ion movement in a tetragonal lattice structure includes jumping to the right spot on the next crystal lattice next to it. Even though the ions are in the same place, they are moving in different directions, which is called anisotropic diffusion. The terms for the lattice are a = b 6¼ c and a = b = c = 90°. Moving along the a- and b-axes is the same and has the same amount of energy because the ion moves the same distance and crosses over similar charge configurations. Moving along the c-axis, on the other hand, needs more space and a different charge arrangement. So, in a tetragonal crystal structure, there are two ways for methylammonium ions to move: P3 along the a- or b-axes, which covers a distance of a units, and P4 along the c-axis, which covers a distance of c units. P3 is better than P4 because it needs to cover less space and has less activation energy.
(Pb2+) ion migration pathways
The Pb4I4 framework is a plane with four Pb2+ ions at the corners and I− ions in the middle of the edges. This is where Pb2+ movement happens. Ions of Pb2+ can move either along the side of the square or across it. P5 along the side of the square plane and P6 along the diagonal are the two ways that Pb2+ could move. High activation energy makes it hard for Pb2+ to move in these ways. Path P5, on the other hand, is better and has less activation energy than Path P6 along the diagonal. In both tetragonal and cubic perovskite crystal lattices, these two movement paths can be found.
Experimental Evidences of Ionic Transport
Experiments can show that ions can move through perovskite materials, and ionic conductivity is very different from electronic conductivity. The activation energy (Ea) ranges from a few eV to hundreds of meV, based on the ionic species, the routes of movement, the activation of light, and the temperature growth of the lattice. This chapter talks about two kinds of device shape that help us understand how ions move and how that changes the structure of the device.
Charge Transport Dynamics in Organolead Halide
Perovskite Materials (Metal/Perovskite/Metal Geometry)
The research is mostly about how ions move and move through the perovskite channel when there is no Electron Transport Layer (ETL) or Holojunction (HTL). The reason for this is that there is no internal moving force, like an electric field built into the interface, and there is no heterojunction made when ions leave or gather at the metal/perovskite/metal interface. To study how charges move through the perovskite active layer, something from the outside, like bias, light, or temperature, needs to be changed.
Organolead halide perovskite materials, such as CH3NH3PbI3, often have flaws like vacancies (VMA, VPb, and VI) and interstitials (MAI, PbI, IMA, and IPb), which are easy to form. In hybrid perovskites, these flaws help with vacancy-mediated ion migration and mixed electronic–ionic conduction by making shallow donor and acceptor levels close to the band edge. But these three kinds of gaps don’t all help conductivity the same amount because they have different transport activation energies.
VI migration is thought to be the main type of migration in hybrid perovskites because it has the lowest activation energy barrier. The VMA vacancy doesn’t make a big difference in conduction because its activation energy is about the same as the other two vacancies. However, the VPb vacancy has a much higher activation energy. A diagram is used to show how ions move, and it usually shows a spin-coated polycrystalline perovskite thin film on a base between two symmetric metal probes, ideally Au or Cr/Au, with a channel length of 20 to 50 µm.
Mechanism
It is possible to see how the movement of electrons and ions interacts in a perovskite material that is at room temperature and in the dark. The openings (VMA, VPb, and VI) are spread out in a random way. In later stages, when no outside disturbance is present, the interaction between electric and ionic transport is known.
Stage 0 (0 to t1 seconds)
At this point, there is no outside bias on the channel, the openings are spread out randomly in the film, and there is no force pushing things to move. Ions are all at rest, which stops them from moving and stops photoconductivity. There is no net potential across the channel because there is no applied positive bias and no ion-induced electric field.
Stage I (t1 to t2 seconds)
Moving and stabilising ions can be broken down into three steps: (a) t1 to t1 + d seconds; (b) t1 + d to t1 + d + a; and (c) t1 + d + a to t2 seconds. At this point, an outside force is put on the object at time t1, which moves all charged particles like electrons, holes, and ions. Ions take longer to show conductivity than electrical currents, which can be seen right away in a very short amount of time, d.
In Stage I (a), an outside force is applied at t1, which creates an electric current with the same charge as the electric current. When an external bias is applied for d seconds, the ions begin to slowly move due to the bias. This creates an ionic current that has the same polarity as the electric current. When these negatively charged ions build up at the contact between the perovskite and electrode, they create an electric field that goes against the direction of the applied electric field. This opposite electric field wipes out some of the applied electric field. This lowers the net electric field across the channel, which makes the electrical current go down.
Stage I (c) happens when the buildup of ions at the contact hits equilibrium and becomes overloaded, mostly because there are no more empty spaces for mobile ions to move. When the balance point is reached, the ion-induced electric field stops growing, and the net electric field across the contact stays the same. The total current going through the device stays the same and doesn’t go down any further. This current is caused by the interaction between the electric current caused by the bias and the current caused by ions.
Stage II (after t2 seconds)
Ion migration in an organolead halide perovskite device can be broken down into three stages: (a) t2 to t2 + dʉ seconds; (b) t2 + dʲ to t2 + dʲ + aʲ seconds; and (c) t2 + dʲ + aʲ seconds after that. In step II (a), the outside bias is taken away, which makes an electric field across the channel caused by the ions. This changes the direction of the net electric field and the current flowing through the gadget right away. The net current is just the electronic current caused by the electric field of the ions. This current at the moment is called I2.
After Stage I (a), the ions that have built up move away from the perovskite-electrode interface. This causes the ion-induced electric field to slowly weaken and the net current across the channel to keep decreasing. In step II(c), the ion-induced electric field is lowered to zero. This means that the device has no current at all. Li et al. studied how current changes when there is an outside bias, and Fig. 4 shows how the current curves of the thin-film perovskite device change over time.
Quantification
Dehui Li and his team measured how much electronic transport and ionic conduction contribute to organolead halide perovskite devices by looking at the current response over time in stage I (with external bias applied) and stage II (without external bias applied). The bi-exponential function was used to fit the fleeting reaction at both steps.
For stage I, the bi-exponential function that fits is written as I°tÚ ¿ I10 þ ð Þ Ie ~ I1 exp ~t ~ t1 te ~ þ ð Þ I1 ~ I10 exp ~t ~ t1 ~ d00 s1 ~ ð2Þ. The same bi-exponential function can be used to fit the time response curve for stage II: IðtÞ = I20 þ ð Þ Ie ~ I2 exp {t ~ t2 se ~ þ ð Þ I2 ~ I20 exp {t ~ t2 ~ ð3.
Experiments are used to measure these factors and get information on how electrons and ions move. Since ion transport is what causes the current to drop, the decay time values s1 and s2 can be used to figure out how ion transport works.
D. Li also worked on the study, which is explained in the next parts.
Bias Dependent
The research is mostly about how ions move through a perovskite pathway. When an external bias is applied, more movable electrons and holes move faster towards the matching electrode. This causes the instant current I1 to rise due to the applied bias and the ion-induced electric field. But this rise only goes up to a certain point. Once it hits a certain number, it stops going up and stops saturating. This might be because there aren’t enough empty spaces for mobile ions to move and build up.
The work by the D. Li group backs up this behaviour when external bias is used. The applied external bias (Vsd) has a straight line connection with the bias-induced electronic current I1. This shows that Ohmic contacts are being made between the metal electrodes and the perovskite thin films. However, once the device is in balance, the steady total current behaves in a way that is not linear. This is because the ion-induced electric field changes as the bias is increased.
In the example given, I1 is greater than I10, and I10 is less than I1. This is because I1 is caused by the ion-induced electric potential, which flows in the opposite direction of I10. When the external bias is taken away at step II, this current is the same as the instant current I2, which is also the electronic current caused by the ion-induced electric potential but with the opposite polarity.
It is about 1.2 V at its highest point across the 20-µm-wide device that is affected by ions. In the same way, I2 first goes up as Vsd goes up and then stops growing at the same saturation current (±4.5 pA) and voltage (±1.2 V). The fact that I2 and I1–I10 have the same saturation behaviour and the same saturation current and voltage proves that the ionic current has almost no effect on the total current.
To figure out how ionic transport works, you have to look at the decay rate k, which is given by s−1 k1 ¿ s^1; k2 ¿ s^2. As predicted for stage I, when an external bias is introduced that is greater, the ions move faster and reach balance. In stage II, on the other hand, the ions that have built up don’t depend on diffusion as much to reach a stable equilibrium state. As the bias for stage II goes up, the decay rate k2 goes down. But when the ion-induced electric field reaches saturation, the ions stop moving towards the opposite electrodes and the distribution of polarised ions reaches a stable state that doesn’t change. This means that the restoration rate k2 stays the same.
Illumination Dependent
Lighting up perovskite materials and devices can speed up the movement of ions, which can damage the devices. Instead of high mobility electrons moving in a band-like pattern, ions move by hopping to nearby sites with different activation energies when the light intensity changes. Increasing the strength of the light significantly lowers the activation energy, which speeds up the movement of ions and makes the device less stable. You can understand this process by looking at stages 0 and I and putting an outside bias across the thin film of perovskite material in the side structure of the device, especially Au/CH3NH3PbI3/Au.
Once the I− and MA+ ions get to their proper electrodes, the cathode goes through a process of I− reduction and then I2 volatilisation while the light supports high-field poling. The MA+ ions move towards the anode and evaporate as CH3NH2. Moisture and light help this process along. This movement of MA+ from the cathode to the anode and evaporation causes CH3NH3PbI3 to slowly change into PbI2.
Y.C. Zhao and his team looked at how the contrast of optical pictures of a perovskite thin film changed when a high electric field was applied along with different amounts of light. In bright light, they saw black lines after 10 seconds of poling extending from the cathode to the anode. In dark light, they didn’t see any change in the contrast in the pictures of the perovskite film caused by ion migration. The fact that the films’ contrast changed when they were exposed to light shows that light makes ion movement easier in perovskites.
Quantification
To find the ionic conductivity (rion) in perovskite sheets, take the mixed conductance and remove the electronic conductance. This is written as rion = rtotal − re. You can find out the total mixed conductivity of the perovskite film by first doing an I-V scan at a speed of 50 Vs−1 in the Au/CH3NH3PbI3/Au device. Then, galvanostatic characterisation, which is the usual way to study mixed conductors, is done with a weak enough current to separate the pure electronic conductivity re from the mixed conductivity rtotal. Ion movement and buildup set the rate, and the electrical conductivity re values are used to find the equilibrium value.
Using the above-mentioned method, Zhao and his team were able to get the pure electronic and ionic conductivities across a wide range of lighting levels and temperatures. The ionisation energy level goes down when there is light because of the blocking effect made by the photo-generated charge carriers. The corresponding ionisation energies for the flaws in the perovskite films are 92, 84, 73, 44, and 53 meV, as seen in optical dynamic images of the film under electric poling and light of different levels in room-temperature air.
The energy barrier Ea affects the formula that shows how ions move in a way that is similar to skipping. The associated curves get much smaller as the lighting level goes up, which shows that the starting energy for ionic transport goes down. The ionic conductance can be broken down into two different linear regions, which are shown in Table 1 as Ea1 (T µ 250 K) and Ea2 (180 < T < 250 K). It is clear that as the light strength rises, the activation energy for both I−/MA+ and H+ ions drops significantly. This fits with the fact that perovskites have better electrical transmission when they are illuminated with high-intensity light. Light speeds up the buildup of ions when the external electric field stays the same. This is because light increases the diffusion coefficient, D D0 exp Ea kBT. So, it’s easier for ions to move when the light strength goes up.
This behaviour of faster ion movement when light is present was also confirmed by D. Li in his research. He did the same experiment in both dark and light conditions by adding an external bias for a set amount of time and removing it across the perovskite material. The ionic transport activation energy was found in the dark and in light by using the current decay rate k2. It was found that the starting energy for ion transport was much lower in the dark (260 meV) than it was in light (80 meV). This shows that light makes the ion movement faster. Ions can move across grain borders more easily when the activation energy for ion migration is low under light.
Finally, figuring out how light affects the movement of ions in perovskite films is very important for learning about these materials’ features. Researchers can learn more about how these materials work and how they might be used in different areas by looking at the ionic conductivity and the screening effect caused by photo-generated charge carriers.
Temperature Dependent
Temperature is a big reason why devices break down because it can change how stable the device is by changing how ions move through it. D. Li and his team looked at how ionic and electronic transport changed over time at different temperatures. They used an outside bias of 3 V to hit the optimum voltage. The fact that I1 and I2 decreased exponentially with falling temperature showed that the electrical current was triggered by temperature.
We used the Arrhenius function to fit these exponential decay plots, which shows that only one type of ions (VI) control the movement of ions in perovskite thin films. Because more than one kind of ionic species could have made a difference, bi- or tri-exponential functions should have been used.
Zhao did a more in-depth study that showed the resistance of the perovskite thin film slowly rising as the ionic conductivity across the channel decreased. The ionic rest time was longer when the temperature was low and got shorter as the temperature went up. As expected, as the temperature went up, the pure electrical resistance went down rapidly, and the flaw ionisation showed up in a clear linear area above 150 K.
A change in conductivity was also seen below the pure electrical resistance. This showed that the motion had an inverted power relationship with temperature. It can also be seen in the movement part of Equation (5). Without photo-excitation, the conductivity flip is not very strong near 100 K. This could be because there are not many carriers near 100 K. Figure 10 shows how the ionic conductivity changes with temperature.
Morphology Dependent
Researchers are still trying to find the best shape for perovskite thin films so that solar cell devices work better and last longer. It has been said that the quality of perovskite thin-film electronics makes them more stable and efficient. Solar cell devices that are more small, even, smooth, and free of pinholes work better and last longer. The main reason for this is that the material in the device has a solid structure, big grains, few grain borders, and a low percentage of empty spaces.
In single crystals with a lot of crystals, the perovskite grains are bigger and there are fewer grain borders. This means that there are fewer empty spaces for ions to move through. This makes the activation energy for ion transport higher in perovskite devices with more crystals. When it comes to less crystalline or polycrystalline perovskite thin films, on the other hand, smaller grains cause bigger grain borders and more flaws in the form of ion gaps. This makes more flaws and ion vacancies than with single crystals, which makes it easier for ions to move around and breaks down the device.
Li and his team did a similar study on the time reaction of nearly single-crystalline microplate devices and polycrystalline perovskite thin films with grains that were about 10 nm in size. The perovskite thin films devices are made using standard spin-coating methods, while the microplate devices are made using vapor-phase intercalation methods that produce grains with bigger sizes and fewer borders between them. Microplates with few flaws have a high solid quality, so there is no dark current that can be seen when the bias Vsd is applied or when the ion-induced potential is set. To study how ionic and electronic charges move in microplates, transient measurements have been taken under light.
Electronic and Ionic Transport Dynamics
in Organolead Halide Perovskites
It takes about 150 meV to start ion movement in microplate devices and 80 meV to start it in perovskite thin-film devices. It’s only 10–25% faster for ions to move through perovskite microplate devices than in thin films. Microplates have a higher ion transport activation energy because they have better crystal quality, bigger grains, fewer grain borders, and fewer empty spaces. In microplate devices, this stops the movement of ions, but it makes it easier for electrons to move. Microplate devices have a low electronic transport activation energy (120–160 meV). This is because there are fewer grain borders, so there is less scattering and charge trapping. The higher ion migration activation energy and lower vacancy concentration make the ion-induced electric field smaller and the hysteresis in the current-voltage curves much smaller. The better crystal quality of the material can greatly lower the movement of ions, lessen or completely get rid of hysteresis, boost the movement of electrons, and make organolead halide perovskite solar cell devices more efficient and stable.
Charge Transport Dynamics in Organolead Halide
Perovskite Solar Cell (Device Geometry)
There is no Electrochemical Transport Mode (ETM) or Holopotential Mode (HTM) in the Au/CH3NH3PbI3/Au device shape, so there is no starting force for ions to move. With perovskite solar cells, on the other hand, the difference in potential between ETM and HTM across the device creates an initial pushing force for charge carriers. There is a potential difference across the device because the electron and hole transport layers are at different potentials. What this difference in potential is equal to is called built-in potential or built-in voltage (Vb). In perovskite, this is the first thing that makes the ions move around.
When there is light, the charges that are created are called photo-carriers or photo-generated charge carriers. When an external forward bias is applied, electrons move towards the ETM and holes move towards the HTM. This charge shift causes photoinduced voltage (VP). This VP moves from ETM to HTM, which is the opposite way of the effective built-in field (Vbi). It tries to release the ions that built up in reaction to the original Vb when it’s dark.
The speed at which ions move through the perovskite material is controlled by how these three forces interact: the effective built-in field (Vbi), the photoinduced voltage (VP), and the applied bias (Vapp). When all of these fields work together, the charges (ions, free electrons, and holes) can move in different ways, which can change how they behave at the surface and in the bulk.
Call resistance (Rs) is the first and most common type of resistance in the path. It is mostly caused by the device’s links to ETM and/or HTM and the measurement equipment itself. Once the charges get into the gadget, their thermodynamics and mechanics tell them to go in different directions. Electrons and holes have a high movement, which means that most of the time they can either pass through or join with other charges. This is called electronic resistance (Relectr). Some of the charges—mostly ions—are stored in the bulk of the perovskite layer and build up there because of the fields that are already there. This makes the bulk capacitive (Cbulk).
The free charges that are still there (electrons and holes) can’t easily move through the device because they are linked to the movement and buildup of ions at the contact. The recombination/transport resistance (Rtr) at the contact is met by these free charges and the ions that are linked to them. After that, these ions and the free charges that go with them either pass through the interface or build up at the interface because of charge trapping and storage, which causes the capacitive behaviour at the interface.
Although ions and their free charges move across the interface, charge and mass also move across the interface because of the ions. Interfacial charge transfer resistance (Rct) is the resistance that the charge transfer meets because the electrode contacts aren’t perfect. This is because of the double-layer capacitance and uneven films.
The movement of these ions changes when outside factors like applied bias, light, and temperature are present. This change in the movement of ions caused by outside disturbances is talked about next.
Bias Dependent
Putting forward bias on perovskite solar cells while they are lit up changes the direction of the applied bias (Vapp) inside the material, which is from ETM to HTM. This extra field causes even more redistribution of ions throughout the material, with Vapp and VP working in the same direction and Vbi working in the opposite way. Depending on how strong the bias Vapp is, the way the net field acts changes.
The net field that acts on the object is shown by Vnet. It is made up of all three of the fields that were talked about above. Depending on how strong these three fields are compared to each other, there are three clear reaction areas that show how ions move through the material: at low, medium, and high Vapp levels.
MA+ ions are at HTM and I ions are at ETM when no bias is applied. This is because of the built-in field (Vb). When a low amount of Vapp is used, the extra effect of VP and Vapp tries to fight with the built-in field (Vb) to release the ions that have gathered. But this extra field isn’t much stronger than the other one, so it doesn’t have a big effect on how much charge builds up. So, the ions stay where they started and don’t move around much. However, as the Vapp grows, the charges that have been collected are screened.
There will be no net directed moving force to cause ion movement after a certain point, when VP + Vapp equals Vbi. If you use middle amounts of Vapp to raise it above this point, the sum of VP and Vapp becomes greater than Vbi, and Vnet goes in the opposite way. This change in the pushing force will cause the ions to move around and gather at the device’s opposite wires again.
At the end of the middle area, all the ions gather at the opposite electrodes, creating the effective built-in potential (Vbi) again, but this time with the opposite polarity. There are positively charged MA+ ions at ETM and negatively charged I ions at HTM. This means that the effective built-in field (Vbi) is now going from ETM to HTM.
When big amounts of Vapp are used, Vnet will grow even more towards VP. If you have a lot of Vapp, Vbi becomes full when all the ions have built up, and Vnet doesn’t go up any further.
Two important points can also be used to describe these three areas: the low level of Vapp, the middle level of Vapp, and the high level of Vapp. When bias is introduced at low and high levels, the trends in the circuit parts are the opposite of each other.
The work is mostly about how ions behave in a circuit, mainly the bulk and double-layer capacitance. As the voltage goes up, there are more flaw sites in the bulk, which means that more ions are moving around and more ions are building up faster at the contact. This causes the depletion width to narrow at the interface, which causes the double-layer capacitance (Cdl) to rise steadily as the bias rises in this area.
When ions move around at the interface, they cause changes in how the ions are spread out in the bulk, which causes the Cbulk to steadily rise. The CDL goes up by *900%, but the Cbulk only goes up by 42%. This is because the applied forward bias (Vapp) has a bigger effect near the interfaces than in the bulk. This means that changes in the Cdl are bigger than changes in the Cbulk.
At the point where VP + Vapp = Vbi, the ions start moving towards the opposite surfaces. This causes the interfacial ion buildup to discharge and the capacitance Cdl to rise at the interface during the middle Vapp region. Once this point is reached, the number of Cdl keeps going down in the middle Vapp area. But the value of Cbulk keeps going up while Cdl is being discharged. This might be because ions that had been gathered at the interface are being discharged and moving towards the other interface, where they will gather in the bulk while the depletion width for Cbulk goes down for a short time as they move towards the other electrode.
When Vapp is high, after the turning point, the value of Cbulk starts going down because the charges have already moved to the other electrodes and built up there. At higher voltage, the value of Cbulk also starts to saturate after hitting its lowest point. This means that the highest point in Cdl is where VP + Vapp = Vbi, and the highest point in Cbulk is where the curve starts to bend.
The study also talks about how ion migration works in two different areas. It says that ions are more mobile when Vapp is low and discharge and move in the opposite way when biases are high. From TW and AW, you can figure out the diffusion coefficient (D) and the ionic conductivity (rion) for moving ions.
Illumination Dependent
Vbi, VP, and Vapp are the fields that move ions around in hybrid perovskite materials. You can’t see the photo-generated field VP when it’s dark, and you can’t move any ions even if you apply bias Vapp. In the dark, EIS plots only show one charge transport regime: the high-frequency spectrum caused by resistance from electronic transport. This is because the forward bias voltage is low. It will not have the low-frequency range that comes from the resistance caused by charge and mass transfer at the contact or Warburg diffusion. This means that only one type of charge transport (electronic) is dominant when it is dark. The low-frequency part of EIS only shows up in the lit perovskite and is mostly linked to the movement of ions.
Ion movement is also affected by the type of light that hits the surface. When three different types of light sources are compared, the low-frequency ion transport component is much lower when white LEDs are used. The white light used doesn’t give off IR or UV waves, which means that ion transport is slower with white LEDs than with solar simulators and IR-only sources. When the perovskite crystal is subjected to white LED alone at 28°C (301 K), the structure grows. However, this change goes the other way when the light is turned off. This ability of gadget degradation to be reversed is talked about in more detail in the next part.
Temperature Dependent
The study uses MAPbI3 samples to look into how ions move through and break down a perovskite device at different temperatures. The activation energy for ion movement goes down as the temperature rises. This makes it easier for ions to move around inside the device because there are more of them. When you heat the perovskite device, the lattice grows, which changes the CH3NH3PbI3 perovskite into PbI2 when it cools down. This change can’t be undone because the MA+ ions that move away from the crystal break their bond with it and evaporate at higher temperatures, leaving behind the lattice. The sample now only has Pb2+ and I− ions, which meet to make PbI2. The following equation can be used to get a rough idea of the activation energy for ion diffusion:
1 TW µ kT h a2a L2 D exp ~Ea kT ~ ð10Ú
It’s possible that the reason for the break in the Arrhenius-like behaviour of ionic transport in CH3NH3PbI3 is the abrupt change in volume caused by a phase transition during lattice expansion. At low temperatures, the activation energy for ion migration is high. At higher temperatures, it drops by a lot, making it easier for ions to move at higher temperatures in CH3NH3PbI3. If you cool the gadget down, you can undo the process.
Size of Alkyl Ammonium Ion Dependent
The size of an ion affects how mobile it is; bigger ions are less mobile. It is harder for FA+ to move into perovskites like MAPbI3, MAXFA1−XPbI3, and FAPbI3. Ionic diffusion coefficients go down as ion size goes up. This means that FAPbI3 and MAXFA1−XPbI3 devices are more stable than MAPbI3 devices. MAXFA1−XPbI3 doesn’t go through structural phase change or lattice expansion, and it doesn’t expand as much at high temperatures as MAPbI3. In mixed systems, the Arrhenius plot shows that ions move continuously, and the activation energy stays the same at all temperatures. The low diffusion rate of FAPbI3 devices makes them steady, which shows a phase change.
Stability Issues
Perovskite-based solar cells have made a lot of progress in how well they work, but they are not stable in direct sunlight. The biggest problem is photoinduced degradation, which stops it from being sold right away. An investigation has been carried out to find out how perovskite-based solar cells break down in natural settings. Ions that can move through the film are often to blame for damage. In the parts that follow, we’ll talk about some of the things that affect ion movement and gadget stability.
Role of Organic Cation
Because the methylammonium ions have less activation energy, methylammonium lead triiodide breaks down faster than formamidinium lead triiodide-based perovskite devices or mixed cationic systems. Ion movement is sped up by lattice expansion and heat contact. When heated up to 55°C, MAPbI3 changes from tetragonal to cubic symmetry. Perovskite solar cells don’t break down as quickly in room light as they do in sunshine or at high temperatures. Changing the methylammonium ions to bigger ones like formamidinium, caesium, or a mix of all three makes perovskite solar cells more stable in dark situations and when they are exposed to light. It’s possible to reverse degradation, but trapping ions at grain borders and ETL or HTL contacts makes the device less efficient over time.
Role of Halide Ion
Lead triiodide-based perovskites have been shown to be the most efficient in the research, but they are not as stable as lead tribromide-based perovskites. Its ionic radius is smaller than that of lead iodide, so its bond is shorter and stronger. In lead triiodide perovskites, this means that the octahedral cavity has less free space. At room temperature, MAPbBr3 has a densely packed cubic crystal structure with a Pm^3m space group. On the other hand, MAPbI3 has a jumbled tetragonal crystal structure with an I4=mcm space group. When the lead-halide bond length is cut down, it takes a lot more energy to move halide ions or organic cations, because there is less empty room for the ions to move. Different groups have found that lowering the lead-halide link length can greatly raise the activation energy of ions.
Role of Crystal Size and Purity
Super-resolution luminescence microspectroscopy is used to see how the photodegradation of perovskite nanocrystals looks. The angle between the lead, halide, and lead affects the optical band gap of perovskite materials. The band gap is smallest when the angle is the widest. The bond angle goes down as the flaw number goes up, which makes the band gap bigger. A link angle of 120° means that the system is totally broken down. Perovskite nanocrystals break down slowly at first, but photoluminescence doesn’t change until the crystals break down quickly. A perovskites structure with three dimensions breaks down into a lead halide structure with two dimensions. The emission spectrum shows a steady change to the blue, with slow decline at first because the crystals don’t have any flaw spots. As the nanocrystal breaks down, more flaws, mostly empty spaces, appear, which makes it more likely that the structure will fall apart quickly.
Conclusion
This part talks about how limiting ion movement in lead halide-based perovskite materials might be able to stop them from breaking down in sunlight. The main cause of photodegradation is ion movement, which can be stopped by decreasing the number of empty ions, making cations bigger, or decreasing the room where ions can move. Several studies have shown ways to make perovskite solar cells more photostable, and a better understanding of how ions move in mixed conductors could lead to their commercial use.
Leave a Reply